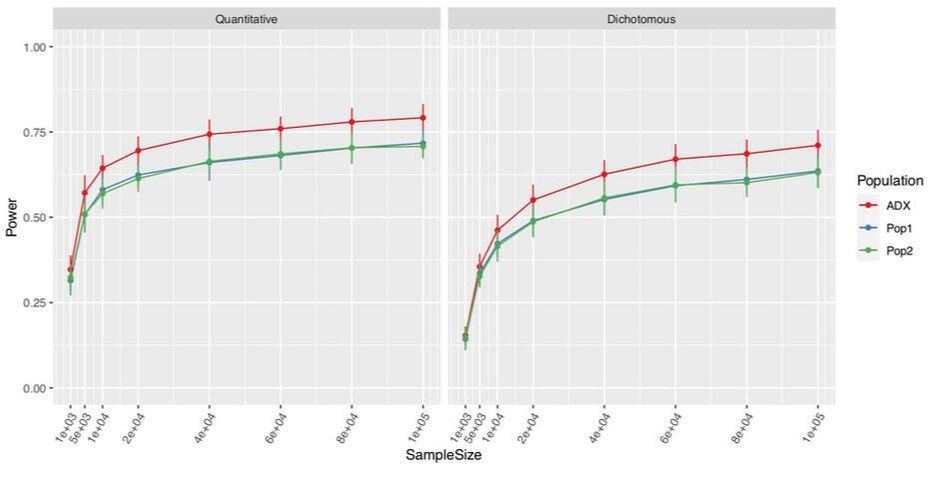
The majority of South Africans have been exposed to M.tb, with a latent tuberculosis infection (LTBI) prevalence of 26% at 5–8 years, 53% at 14–17 years and >75% at 25 years. Population-wide LTBI, high TB incidence, and low HIV infection allow us to capture a large sample size and ideal cases-control ratio (48% cases, 90% HIV-negative, and n=1100 currently collected). Each participant in our study has matched DNA and epidemiological data (smoking behavior, alcohol consumption, comorbidities [HIV, diabetes, asthma], education and SES, and anthropometrics).